Clay-polymer nanocomposites
Over the course of the MAPPER project, we will develop a "bottom-up" multiscale simulation mechanism that will, through its advances, allow the study and design of layered mineral composites in areas having a substantial potential impact such as energy applications (oil industry additives), materials applications (nano composites materials) and biomedical applications (drug delivery). The microscopic structure and mechanisms of layered nanomaterials operate over many different length scales, ranging from nanometers to microns; each length scale needs to be properly simulated to fully understand their features. This knowledge will eventually lead to the design of novel layered mineral systems with properties tailored to their application.
Simulation on the atomic scale can be time consuming; simulations have to compute and tract the position and motion of each individual atom. While this may be suitable for simulations of small molecules, when one wishes to simulate a system containing billions of atoms, the computer time required makes such a study unfeasible. Despite increases in computer power (as described by Moore's Law) and improved efficiency in algorithms, there are many situations where we need to simulate a very large system in atomistic detail, but cannot due to the associated computational overheads. An answer to such a bottleneck is to reduce the degrees of freedom in the system by combining several atoms together into a "pseudo"-particle, defined to possess the properties of the atoms it is composed of. By effectively removing the unimportant internal degrees of freedom, we can speed up simulation times by a factor of 100 or even 1000, yet still retain the required accuracy of a full atomistic simulation. These coarse-grained simulations can push atomistic techniques to the macro level, in terms of both length (micron instead of nanometer) and time (microsecond rather than nanosecond), allowing one to study previously unavailable phenomena, such as long-time diffusion and large-scale structure.
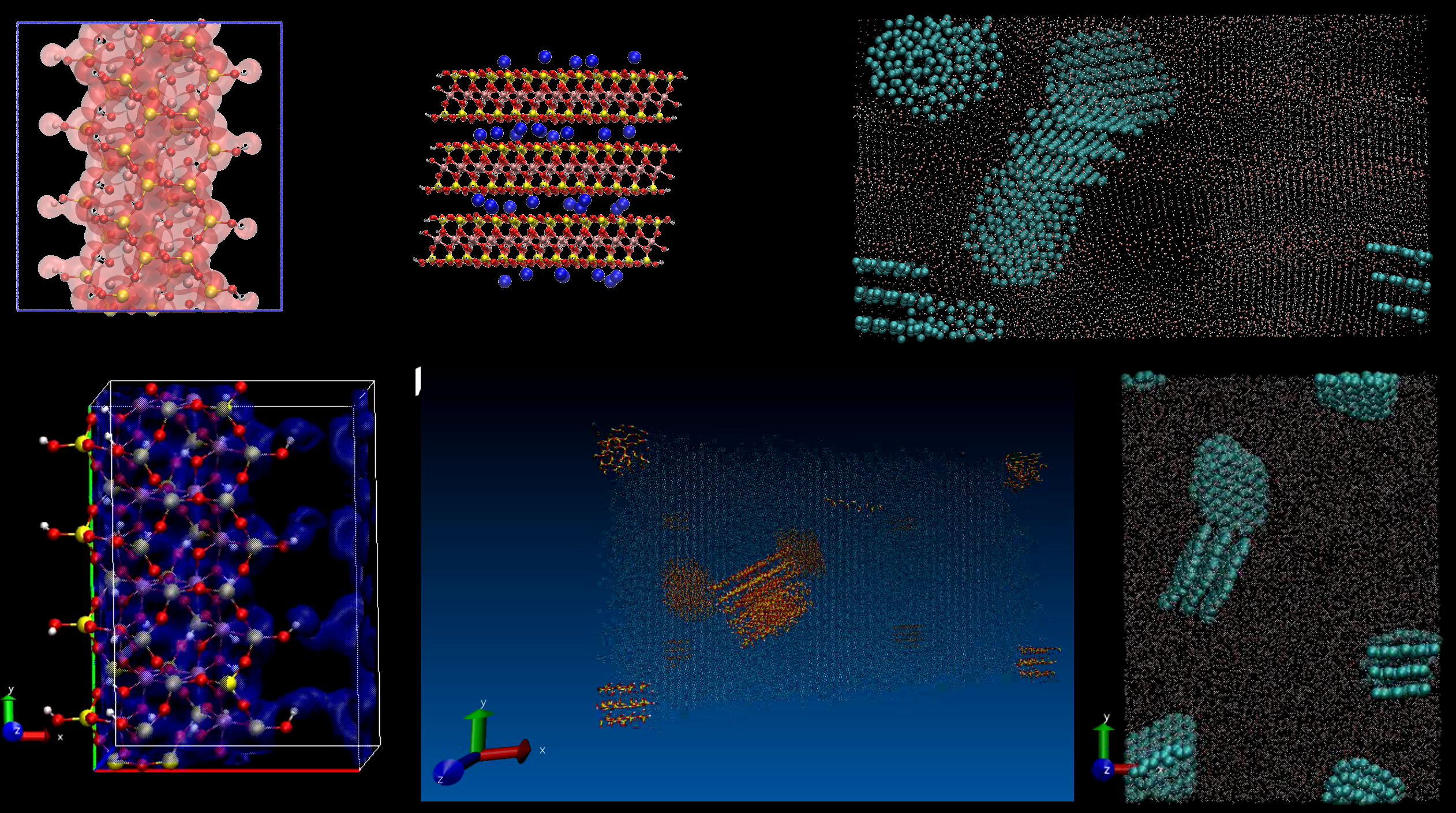
Figure 1: Various renderings from the clay-polymer nanocomposites application.
Such techniques are becoming more and more commonplace in the biological and polymer science domain. However, problems remain; firstly, how do you split up your atomic system into "pseudo"-particles, and, most importantly, how do you define their properties such that they reproduce those of the atoms that form them? Previous work has side-stepped these problems by creating systems with arbitrary parameters, such that trends could be addressed qualitatively, but no quantitative information can be easily be extracted.
Similarly, bond breaking and formation of atoms requires simulation of the electronic structure. Such techniques are computationally very expensive, yet combining them into a multiscale simulation will allow polymerization mechanisms in layered minerals to be studied and correct parameterisation of classical models to be performed.
We use the MAPPER infrastructure, tools and software to transparently couple these three levels of simulation in a loosely coupled scheme across distributed computing infrastructures. Combined with our scientific advances, this will facilitate the understanding of the underlying mechanisms of layered nanominerals on both the atomic and much larger scales.
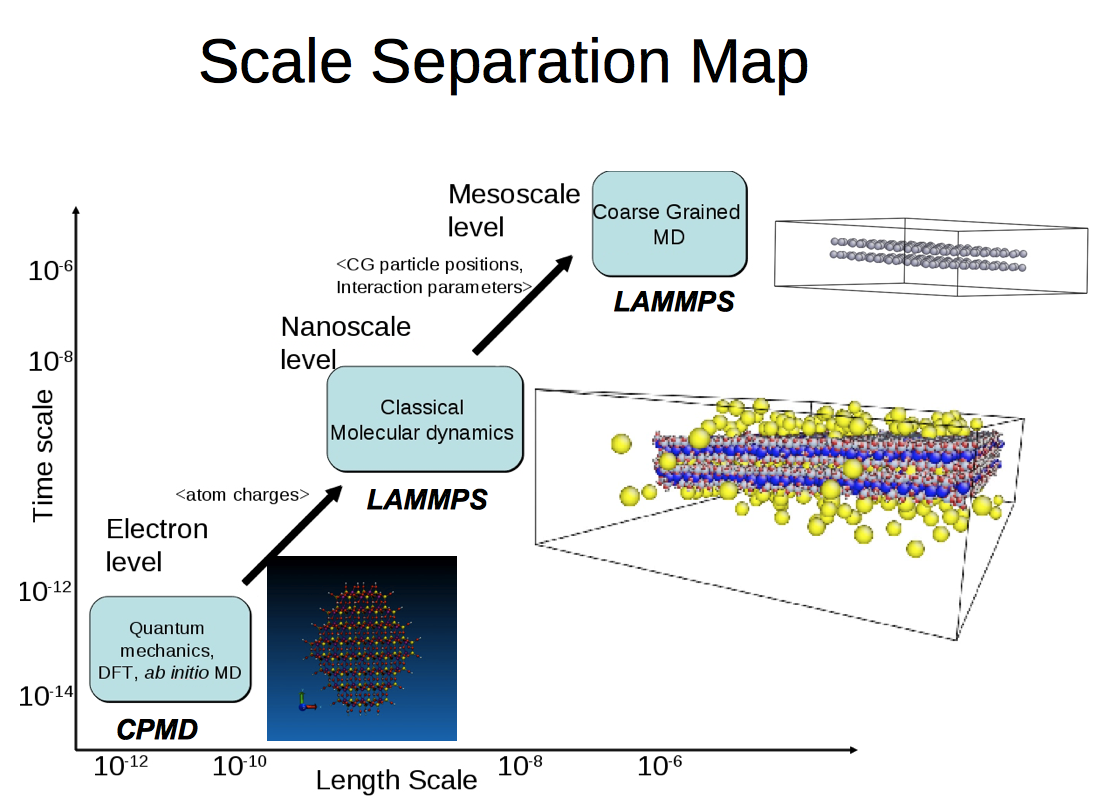
Figure 2: Scale separation map of the clay-polymer interactions application
More information can be found at: http://inside.hlrs.de/htm/Edition_01_12/article_24.html
Relevant papers:
[1] J. Suter, D. Groen, L. Kabalan and P. Coveney: Distributed Multiscale Simulations of Clay-Polymer Nanocomposites, Materials Research Symposium, San Francisco, United States of America, April 2012.
[2] D. Groen, J. Suter, P. Coveney: Modelling Distributed Multiscale Simulation Performance: An Application to Nanocomposites, In Proceedings of The Seventh IEEE International Conference on e-Science Workshops, Stockholm, Sweden, 5-8 December 2011. IEEE Computer Society, Washington, DC, USA, 105 - 111, 2011. doi:10.1109/eScienceW.2011.37